|
Biorithm
1.1
|
|
Biorithm
1.1
|
#include <old-annotations.h>
Classes | |
| class | BioFunctionsConstIterator |
| A class that iterates over all functions of a particular type. It contains the methods hasNext() and next() to sustain the advance. This iterator cannot be used to modify the instance of MyAnnotations on which it is invoked. More... | |
| class | BioFunctionsIterator |
| A class that iterates over all functions of a particular type. It contains the methods hasNext() and next() to sustain the advance. This iterator can be used to modify the instance of MyAnnotations on which it is invoked. More... | |
| class | BioFunctionsIteratorMultiIndex |
| This class implements an iterator over all functions stored in an instance of MyAnnotations. The iterator automatically ranges over all function categories and within each category over all functions belonging to that category. More... | |
Public Member Functions | |
| void | setOverlappingFunctionsFlag () |
| void | addAnnotation (string gene, const BioFunction &function, string evidenceCode, MyGainAnnotationType status=ANNOTATED_STATE, bool msa=false) |
| Add the annotation function for gene. | |
| void | addAnnotation (string gene, string function, string funcType, string evidenceCode, MyGainAnnotationType status=ANNOTATED_STATE, bool msa=false) |
| Add the annotation function for gene in the functional category funcType. | |
| void | applyTruePathRule (GeneOntology &go, bool applyUpward=true, bool applyDownward=true, bool applySideways=true) |
| bool | checkTruePathRule (GeneOntology &go, ostream *ostr=NULL, bool *annotationsOK=NULL, bool *unknownOK=NULL) |
| Returns true if the annotations satisfy the GO true path rule. | |
| void | computeDifference (MyAnnotations &other, MyAnnotations &difference, bool restrictToCommonGenes=false) |
| Computes the annotations that are in the invocant but not in other/. | |
| void | computeEnrichments (const set< string > &geneSet, const GeneOntology *go, vector< EnrichmentRecord< string, string > > &rejalts) |
| Compute the functions in the invocant that are enriched in a set of genes. If go is not NULL, use the Ontologizer algorithm. | |
| void | computeEnrichmentsGenGO (const set< string > &geneSet, vector< EnrichmentRecord< string, string > > &rejalts, const GeneOntology *go=NULL, GenGOParametersOptimisationType optType=GENGO_PARAMETER_OPTIMISATION_NONE) const |
| Compute the functions in the invocant that are enriched in a set of genes using the GenGO algorithm (http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2553574). | |
| void | computeEnrichmentsGenGO (const set< string > &geneSet, string functionType, vector< EnrichmentRecord< string, string > > &rejalts, const GeneOntology *go=NULL, GenGOParametersOptimisationType optType=GENGO_PARAMETER_OPTIMISATION_NONE) const |
| Compute the functions of a specific type in the invocant that are enriched in a set of genes using the GenGO algorithm (http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2553574). | |
| MyNT | computeFunctionFrequency (string funcType, string func) |
| Return the fraction of (annotated) genes that are annotated by func, only considering genes annotated by functions belonging to the category funcType. | |
| void | computeStatistics () |
| void | computeGeneCounts (map< BioFunction, unsigned int > &geneCounts) |
| void | copyAnnotationsForFunctions (const set< BioFunction > &functions, MyAnnotations &annotations) |
| virtual MyAnnotations::BioFunctionsIterator | functions (string type) |
| virtual MyAnnotations::BioFunctionsIteratorMultiIndex | functionsMultiIndex () const |
| set< string > | functionTypes () const |
| Return a set containing the different function types (categories). Example of function categories are "cellular
component", "molecular function", and "biological process" in the Gene Ontology. | |
| MyNT | getAnnotatedFunctionWeight (string gene, string func, string type) |
| Return the weight of the evidence code of the annotation of gene by function func. | |
| MyNT | getAnnotatedFunctionWeightProbabilisticOr (string gene, string func, string type) |
| Return the weight of the evidence code of the annotation of gene by function func, after combining multiple evidence code weights using the "probabilistic or". | |
| map< string, MyNT > | getAnnotatedFunctionWeights (string gene, string func, string type) |
| Return the weights of the evidence codes for the annotation of gene by function func. | |
| map< string, MyNT > | getAnnotatedWeights (string gene, string type) |
| Return the weights of the evidence codes for the annotation of gene by all functions in the category type. | |
| void | getAnnotatedGenes (string functype, set< string > &annotatedGenes) |
| Return the set of all genes annotated with a given function type. If the given function type is the empty string, return all annotated genes. | |
| void | getAnnotations (string type, map< string, set< string > > &annotations) const |
| Return the set of all annotations for functions of a given type, keyed by function. | |
| void | getAnnotationsByGene (string type, map< string, set< string > > &annotations) const |
| Return the set of all annotations for functions of a given type, keyed by gene. | |
| virtual void | getAnnotations (string type, string function, set< string > &annotations) const |
| void | getAnnotations (const GOFunction *function, set< string > &annotations) const |
| void | getAnnotations (const set< GOFunction * > &functions, set< string > &annotations) const |
| virtual void | getAnnotationsForGene (string type, string gene, set< string > &annotations) const |
| void | getEvidenceCodeCounts (map< string, unsigned int > &ecCounts) const |
| Return the number of times each evidence code appears among all the annotations. | |
| void | getEvidenceCodeCounts (string funcType, string funcId, map< string, unsigned int > &ecCounts) const |
| Return the number of times each evidence code appears among the annotation for a particular function. | |
| MyNT | getFunctionFrequency (string function) const |
| Return the fraction of genes annotated by a function. | |
| string | getFunctionType (string functionName) const |
| Determine function's type from its name. | |
| bool | hasExperimentallyAnnotatedFunction (string gene, string category) const |
| Return true if and only if the gene is annotated via an experimental evidence code by any function in the specified category. | |
| bool | hasAnnotatedFunction (string gene) const |
| Return true if and only if gene is annotated with at least one function (in any category). | |
| bool | hasAnnotatedFunction (string gene, string category) const |
| Return true if and only if gene is annotated with at least one function from the specified functional category. | |
| bool | isNotKnownToBeAnnotatedWithFunction (string gene, string func, string type) |
| Return true if and only if gene is not annotated with respect to the function func belonging to functional category type. | |
| bool | isNotKnownToBeAnnotatedWithFunctionSansTruePathRule (string gene, string func, string type, GeneOntology &goDAG) |
| Return true if and only if gene is not annotated with respect to the function func belonging to functional category type. | |
| bool | hasAnnotatedGene (string func, string type) const |
| Return true if and only if function (belong to functional category type) annotates at least one gene. | |
| bool | hasAnnotatedGene (const BioFunction &function) const |
| Return true if and only if function annotates at least one gene. | |
| bool | haveSameFunction (string gene1, string gene2, const BioFunction &function) |
| bool | haveSameFunction (string gene1, string gene2, const vector< BioFunction > &functionVector) |
| template<class InputIterator > | |
| bool | haveSameFunction (string gene1, string gene2, InputIterator first, InputIterator last) |
| bool | haveSameFunction (string gene1, string gene2, string type) |
| bool | haveSameFunction (string gene1, string gene2) |
| bool | haveOverlappingFunction (string gene1, string function, string type) |
| bool | isAnnotatedWithFunction (string gene, string func, string type, bool beforeTruePathRule=false) |
| Return true if and only if gene is annotated with the function func belonging to the function category type. | |
| bool | isAnnotatedWithFunctionExperimentally (string gene, string func, string type) |
| Return true if and only if gene has the function func belonging to the function cateogry type and if the evidence code for the annotation is experimental, e.g., IDA, IPI, IEP, TAS, or NAS. | |
| bool | isUnknownFunction (string func, string type) const |
| Return true if and only if func is the unknown function for function category "type". | |
| void | keepAnnotationsForGenes (set< string > &geneSet) |
| unsigned int | numAnnotations (string category="") |
| Return the total number of annotations, i.e., gene-function pairs where the function annotates the gene. | |
| unsigned int | numUnknownAnnotations (string category="") |
| Return the total number of non-annotations, i.e., gene-function pairs where the annotation status of the gene with respect to the function is unknown. | |
| unsigned int | numAnnotatedGenes (string funcType, string function) |
| unsigned int | numAnnotatedGenes (string funcType="") |
| Return the total number of genes annotated by any function in the category funcType. | |
| unsigned int | numAnnotatingFunctions (string gene, string funcType) |
| Return the number of functions (of category funcType) annotating gene. | |
| unsigned int | numAnnotatingFunctions (string funcType="") |
| Return the total number of functions (of category funcType) annotating at least one gene. | |
| unsigned int | numNonAnnotatedGenes (string funcType) |
| Return the number of genes not annotated by any function in the category funcType. | |
| unsigned int | numTotalNonAnnotatedGenes (string function, string funcType) |
| Return the total number of genes not annotated by the function (these genes have unknown status), including those genes not annotated by any function in the category funcType. | |
| unsigned int | numNonAnnotatedGenes (string funcType, string function) |
| unsigned int | numUnknownAnnotatingFunctions (string gene, string funcType) |
| Return the number of functions in the category funcType that do not annotate gene. | |
| unsigned int | numNegativeGenes (string funcType, string function) |
| void | print (string fileName) |
| Print annotations to a file. | |
| void | printWithName (string fileName) |
| void | printNumAnnotationsPerFunction (ostream &numFunStream) |
| Print information on the number of annotations, depth, and category for each function. | |
| void | read (string fileName, string dataType="unweighted", const map< string, set< string > > *nodeAliases=NULL, bool mostSpecificAnnotations=false, const set< string > *evidenceCodesToIgnore=NULL) |
| void | read (string fileName, unsigned int keySize) |
| void | readGO (string fileName, unsigned int altSymbolCol=0) |
| void | readEvidenceCodeWeights (string ecwFile) |
| Read evidence code weights. See GOEvidenceCodes::readEvidenceCodeWeights() for details. | |
| void | subtract (MyAnnotations &other, MyAnnotations &difference) |
| Find the gene-function annotation pairs in the invocant that are not in other and store these pairs in difference. | |
| void | storeGODAG (GeneOntology &go) |
| void | readOverlappingFunctions (string fileName) |
Friends | |
| class | NewBioFunctionsIterator |
This class stores (gene, function) pairs, where the function annotates the gene.
Here is an example of creating an instance of MyAnnotations:
GeneOntology go;
go.read("gene-ontology.obo");
MyAnnotations ann;
ann.read("annotations.txt");
ann.applyTruePathRule(go, true, false, false);
One common use of this class is to loop through all functions stored in it. Here is a skeleton of a typical loop through a MyAnnotations object called annotations. Further more, in this skeleton we also show how to get all the genes annotated by the function.
set< string > types = annotations.functionTypes();
set< string >::const_iterator titr;
for (titr = types.begin(); titr != types.end(); titr++)
{
MyAnnotations::BioFunctionsIterator bfItr = annotations.functions(*titr);
while (bfItr.hasNext())
{
BioFunction function(*titr, bfItr.next());
set< MyNodeId > genesAnnotatedByFunction;
annotations.getAnnotations(function.getCategory(), function.getId(), genesAnnotatedByFunction);
doSomething(function);
doSomethingElse(genesAnnotatedByFunction);
}
}
Another way to loop over all the functions is the following (this method will replace the previous one at some point, with MyAnnotations::functionsMultiIndex() being renamed to MyAnnotations::functions()):
MyAnnotations::BioFunctionsIteratorMultiIndex mitr = annotations.functionsMultiIndex();
BioFunction function;
while (mitr.hasNext())
{
function = mitr.next();
process the function.
}
| void MyAnnotations::applyTruePathRule | ( | GeneOntology & | go, |
| bool | applyUpward = true, |
||
| bool | applyDownward = true, |
||
| bool | applySideways = true |
||
| ) |
Transfer annotations up and down the Gene Ontology DAG.
In addition to go, the method takes three optional boolean parameters that default to true. These parameters control in which "direction" in the Gene Ontology DAG, the method should apply the true path rule.
| [in] | go,a | reference to an instance of GeneOntology. |
| [in] | applyUpward; | if this boolean is true, for every gene g annotated by a function f, ensure that every ancestor of f also annotates g. |
| [in] | applyDownward; | if this boolean is true, for every gene g annotated by a function f, ensure that the annotation status of g with respect to every descendant of f is unknown. The method only considers a pair (g, f) only if f is one of the most specific annotations of g, i.e., if no descendant of f annotates g. |
| [in] | applySideways; | if this boolean is true, for every gene g annotated by a function f (when no descendant of f annotates g), ensure that the annotation status of g with respect to f' is unknown, for every function f' that satisfies the following criteria: |
(a) f' is a descendant of an ancestor of f and
(b) f' is an ancestor of a descendant of f.
| bool MyAnnotations::checkTruePathRule | ( | GeneOntology & | go, |
| ostream * | ostr = NULL, |
||
| bool * | annotationsOK = NULL, |
||
| bool * | unknownOK = NULL |
||
| ) |
Returns true if the annotations satisfy the GO true path rule.
| [in] | go,a | reference to an instance of GeneOntology. |
| [in] | ostr,a | pointer to an output stream. If this parameter is not NULL, the method prints details of violating annotations to the stream. |
| inout] | annotationsOK, a pointer to a bool whose default value is NULL. If the pointer is not NULL, the method checks if the annotations have been correctly transferred upward. The method also sets the value of the boolean pointed to to true if the transfer is correct. | |
| inout] | unknownOK, a pointer to a bool whose default value is NULL. If the pointer is not NULL, the method checks if the annotations have been correctly transferred downward. The method also sets the value of the boolean pointed to to true if the transfer is correct. |
| void MyAnnotations::computeDifference | ( | MyAnnotations & | other, |
| MyAnnotations & | difference, | ||
| bool | restrictToCommonGenes = false |
||
| ) |
Computes the annotations that are in the invocant but not in other/.
This method computes the set difference between two instances of MyAnnotations. The result is an instance of MyAnnotations contains every gene-function pair that is in the invocant but not in other.
| [in] | rhs,an | instance of MyAnnotations. |
| [out] | difference,an | instance of MyAnnotations that will hold the result. |
| [in] | restrictToCommonGenes,if | true, difference will only contain annotations for genes that have annotations in other too. The default value is true. |
| void MyAnnotations::computeEnrichments | ( | const set< string > & | geneSet, |
| const GeneOntology * | go, | ||
| vector< EnrichmentRecord< string, string > > & | rejalts | ||
| ) |
Compute the functions in the invocant that are enriched in a set of genes. If go is not NULL, use the Ontologizer algorithm.
| void MyAnnotations::computeEnrichmentsGenGO | ( | const set< string > & | geneSet, |
| vector< EnrichmentRecord< string, string > > & | rejalts, | ||
| const GeneOntology * | go = NULL, |
||
| GenGOParametersOptimisationType | optType = GENGO_PARAMETER_OPTIMISATION_NONE |
||
| ) | const |
Compute the functions in the invocant that are enriched in a set of genes using the GenGO algorithm (http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2553574).
| [in] | geneSet,the | set of genes for which the set of enriched functions must be computed. |
| [out] | rejalts,a | vector holding the enriched functions. |
| [in] | go,a | pointer to an instance of GeneOntology. If go is not NULL, use it to speed up the algorithm. |
| void MyAnnotations::computeEnrichmentsGenGO | ( | const set< string > & | geneSet, |
| string | functionType, | ||
| vector< EnrichmentRecord< string, string > > & | rejalts, | ||
| const GeneOntology * | go = NULL, |
||
| GenGOParametersOptimisationType | optType = GENGO_PARAMETER_OPTIMISATION_NONE |
||
| ) | const |
Compute the functions of a specific type in the invocant that are enriched in a set of genes using the GenGO algorithm (http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2553574).
| [in] | functionType,a | string specifying the category/type of the functions to consider (e.g., MSigDB_correlation, KEGG, or "biological_process"). |
| virtual MyAnnotations::BioFunctionsIterator MyAnnotations::functions | ( | string | type | ) | [inline, virtual] |
Return an iterator over the functions of a particular type.
| [in] | type,a | function "type" (e.g., KEGG, c, f, or p, where "c", "f", and "p" stand for GO's cellular component, molecular function, and biological process categories). |
| virtual MyAnnotations::BioFunctionsIteratorMultiIndex MyAnnotations::functionsMultiIndex | ( | ) | const [inline, virtual] |
Return an iterator over the functions of a particular type using the multi-index data structure.
| set< string > MyAnnotations::functionTypes | ( | ) | const [inline] |
Return a set containing the different function types (categories). Example of function categories are "cellular component", "molecular function", and "biological process" in the Gene Ontology.
| MyNT MyAnnotations::getAnnotatedFunctionWeight | ( | string | gene, |
| string | func, | ||
| string | type | ||
| ) |
Return the weight of the evidence code of the annotation of gene by function func.
| [in] | gene | a string denoting the ID of the gene. |
| [in] | func | a string denoting the ID of the function. |
| [in] | type | a string denoting the type of function of interest, e.g., "b" for GO biological process or "biocarta" for Biocarta pathways. |
| MyNT MyAnnotations::getAnnotatedFunctionWeightProbabilisticOr | ( | string | gene, |
| string | func, | ||
| string | type | ||
| ) |
Return the weight of the evidence code of the annotation of gene by function func, after combining multiple evidence code weights using the "probabilistic or".
See MyAnnotations::getAnnotatedFunctionWeight() for documentation on the parameters. If there is more than one evidence code for the annotation of gene by func, then the method combines the weights of these evidence codes using the "probabilistic or": for two weights 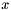 and
and 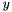 , their probabilistic OR is
, their probabilistic OR is 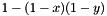 . This formula is straightforward to generalize to multiple weights.
. This formula is straightforward to generalize to multiple weights.
| map< string, MyNT > MyAnnotations::getAnnotatedFunctionWeights | ( | string | gene, |
| string | func, | ||
| string | type | ||
| ) |
Return the weights of the evidence codes for the annotation of gene by function func.
| [in] | gene | a string denoting the ID of the gene. |
| [in] | func | a string denoting the ID of the function. |
| [in] | type | a string denoting the type of function of interest, e.g., "b" for GO biological process or "biocarta" for Biocarta pathways. |
| void MyAnnotations::getAnnotatedGenes | ( | string | functype, |
| set< string > & | annotatedGenes | ||
| ) |
Return the set of all genes annotated with a given function type. If the given function type is the empty string, return all annotated genes.
| [in] | functype,a | string denoting the type of function of interest. |
| [out] | annotatedGenes,a | set in which to store the genes annotated with the given function type. |
| map< string, MyNT > MyAnnotations::getAnnotatedWeights | ( | string | gene, |
| string | type | ||
| ) |
Return the weights of the evidence codes for the annotation of gene by all functions in the category type.
| [in] | gene | a string denoting the ID of the gene. |
| [in] | type | a string denoting the type of function of interest, e.g., "b" for GO biological process or "biocarta" for Biocarta pathways. |
| void MyAnnotations::getAnnotations | ( | string | type, |
| map< string, set< string > > & | annotations | ||
| ) | const |
Return the set of all annotations for functions of a given type, keyed by function.
| [in] | type,a | string denoting the type of function of interest, e.g., "b" for GO biological process or "biocarta" for Biocarta pathways. |
| [out] | annotations,a | map, each of whose keys is a function and each of whose values is a set of genes annotated with the function. |
| void MyAnnotations::getAnnotations | ( | string | type, |
| string | function, | ||
| set< string > & | annotations | ||
| ) | const [virtual] |
Return the set of all annotations for a given function.
| [in] | type,a | string denoting the type of function of interest, e.g., "b" for GO biological process or "biocarta" for Biocarta pathways. |
| [in] | function,a | string denoting the ID of the function. |
| [out] | annotations,a | set of genes annotated with the function. |
| void MyAnnotations::getAnnotations | ( | const GOFunction * | function, |
| set< string > & | annotations | ||
| ) | const |
Return the set of all annotations for a given function.
| [in] | function,a | GOFunction pointer to the function of interest |
| [out] | annotations,a | set of genes annotated with the function. |
| void MyAnnotations::getAnnotations | ( | const set< GOFunction * > & | functions, |
| set< string > & | annotations | ||
| ) | const |
Return the set of all annotations for a set of functions, all of the same type.
| [in] | functions,a | set containing GOFunction pointers. |
| [out] | annotations,a | set of genes annotated with the function. |
| void MyAnnotations::getAnnotationsByGene | ( | string | type, |
| map< string, set< string > > & | annotations | ||
| ) | const |
Return the set of all annotations for functions of a given type, keyed by gene.
| [in] | type,a | string denoting the type of function of interest, e.g., "b" for GO biological process or "biocarta" for Biocarta pathways. |
| [out] | annotations,a | map, each of whose keys is a gene and each of whose values is a set of functions annotating that gene. |
| void MyAnnotations::getAnnotationsForGene | ( | string | type, |
| string | gene, | ||
| set< string > & | annotations | ||
| ) | const [virtual] |
Return the set of all annotations for a given gene.
| [in] | type,a | string denoting the type of functions of interest, e.g., "b" for GO biological process or "biocarta" for Biocarta pathways. If this parameter is the empty string, the method returns all functions annotating the gene. |
| [in] | gene,a | string denoting the ID of the gene. |
| [out] | annotations,a | set of functions annotating the gene. function. |
| void MyAnnotations::getEvidenceCodeCounts | ( | map< string, unsigned int > & | ecCounts | ) | const [inline] |
Return the number of times each evidence code appears among all the annotations.
| [out] | ecCounts | a map each of whose keys is an evidence code and whose value is the number of times that evidence code appears in the annotations. |
| void MyAnnotations::getEvidenceCodeCounts | ( | string | funcType, |
| string | funcId, | ||
| map< string, unsigned int > & | ecCounts | ||
| ) | const |
Return the number of times each evidence code appears among the annotation for a particular function.
| [in] | funcType | the category the function belongs to. |
| [in] | funcId | the id of the function. |
| [out] | ecCounts | a map each of whose keys is an evidence code and whose value is the number of times that evidence code // return true if gene1 and gene2 have the given function. bool haveSameFunction(string gene1, string gene2, const BioFunction &function); appears in the annotation for the function specified by the first two arguments. |
| string MyAnnotations::getFunctionType | ( | string | functionName | ) | const |
Determine function's type from its name.
The method assumes that there is only one type that any function can have, so it returns the first matching type.
| [in] | functionName,the | name of the function. |
| bool MyAnnotations::isAnnotatedWithFunction | ( | string | gene, |
| string | func, | ||
| string | type, | ||
| bool | beforeTruePathRule = false |
||
| ) |
Return true if and only if gene is annotated with the function func belonging to the function category type.
| [in] | gene | the identifier of the gene |
| [in] | func | the identifier of the function |
| [in] | type | the category that func belongs to |
| [in] | beforeTruePathRule | If this Boolean parameter is true, the method will return true iff gene is annotated with func and if this annotation existed before MyAnnotations::applyTruePathRule() was invoked. This parameter is used by MyAnnotations::isNotKnownToBeAnnotatedWithFunctionSansTruePathRule(). |
| bool MyAnnotations::isNotKnownToBeAnnotatedWithFunction | ( | string | gene, |
| string | func, | ||
| string | type | ||
| ) | [inline] |
Return true if and only if gene is not annotated with respect to the function func belonging to functional category type.
| [in] | gene | the identifier of the gene |
| [in] | func | the identifier of the function |
| [in] | type | the category that func belongs to |
The method returns true if (i) the gene is not annotated with any function in the functional category type or (ii) this gene is in the list of genes not known to be annotated with this function (i.e., the annotation status of this gene is 0 with respect to this function.)
| bool MyAnnotations::isNotKnownToBeAnnotatedWithFunctionSansTruePathRule | ( | string | gene, |
| string | func, | ||
| string | type, | ||
| GeneOntology & | goDAG | ||
| ) |
Return true if and only if gene is not annotated with respect to the function func belonging to functional category type.
| [in] | gene | the identifier of the gene |
| [in] | func | the identifier of the function |
| [in] | type | the category that func belongs to |
| [in] | goDAG | an instance of GeneOntology |
The method returns true if (i) the gene is not annotated with any function in the functional category type or (ii) this gene is in the list of genes not known to be annotated with this function (i.e., the annotation status of this gene is 0 with respect to this function.)
The difference between this method and MyAnnotations::isNotKnownToBeAnnotatedWithFunction() is that this method should be invoked when MyAnnotations::applyTruePathRule() has not been called with the applyDownward argument set to true. This method explicitly checks the ancestors of func in goDAG to check if any of them are the most specific annotation for gene.
| bool MyAnnotations::isUnknownFunction | ( | string | func, |
| string | type | ||
| ) | const [inline] |
Return true if and only if func is the unknown function for function category "type".
| void MyAnnotations::keepAnnotationsForGenes | ( | set< string > & | geneSet | ) |
Delete annotations whose function is not in functionSet
| functionSet,a | reference to a set of function identifiers. TODO: THIS DOESN'T WORK! (but it would be useful) Delete annotations for genes not in geneSet. |
| geneSet,a | reference to a set of gene identifiers. |
| unsigned int MyAnnotations::numAnnotatedGenes | ( | string | funcType, |
| string | function | ||
| ) | [inline] |
Return the number of genes annotated by the function (of category funcType).
| unsigned int MyAnnotations::numAnnotatingFunctions | ( | string | funcType = "" | ) | [inline] |
Return the total number of functions (of category funcType) annotating at least one gene.
| [in] | funcType,the | functional category of interest. If this parameter is the empty string (which is the default value), the method returns the total number of functions annotating at least one gene, summed over all categories. |
| void MyAnnotations::printWithName | ( | string | fileName | ) |
Same as MyAnnotations::print(string) except also print an additional column with the goname for the given goid. WARNING: This should only be used if the annotations are GO annotations and the GO DAG is known.
| void MyAnnotations::read | ( | string | fileName, |
| string | dataType = "unweighted", |
||
| const map< string, set< string > > * | nodeAliases = NULL, |
||
| bool | mostSpecificAnnotations = false, |
||
| const set< string > * | evidenceCodesToIgnore = NULL |
||
| ) |
Read annotations from a file.
| [in] | fileName,the | name of the file containing the annotations. |
| [in] | dataType,the | type of the file. Ignore this parameter. It will go away in the future. |
| [in] | nodeAliases,a | map from gene identifiers in fileName to another namespace. Each gene may have multiple aliases. After reading each (gene, function) pair from fileName, the method stores function as an annotation for each alias of gene. The method does not store the original annotation. |
| [in] | mostSpecificAnnotations,a | boolean. If the parameter is true, the method assumes that the annotations in fileName are only the most specific annotations, e.g., with respect to the GO DAG. |
| [in] | evidenceCodesToIgnore,a | pointer to a set whose elements are evidence codes that the method should ignore. The method will not store annotations with these evidence codes. |
| void MyAnnotations::read | ( | string | fileName, |
| unsigned int | keySize | ||
| ) |
Read annotations from a file.
| [in] | fileName,the | name of the file containing the annotations. |
| [in] | keySize,the | number of columns that identify an annotated object uniquely. If this number is 1, then column 0 contains the identifier (for a gene, for example). If this number is 2, then columns 0 and 1 contain the identifier (for an interaction, for example). And so on ... |
The method assumes that after columns [0 .. keySize) come the id of the annotating function, the category of the annotating function, an optional annotation status (which is always 1), and an optional evidence code.
| void MyAnnotations::readEvidenceCodeWeights | ( | string | ecwFile | ) | [inline] |
Read evidence code weights. See GOEvidenceCodes::readEvidenceCodeWeights() for details.
| void MyAnnotations::readGO | ( | string | fileName, |
| unsigned int | altSymbolCol = 0 |
||
| ) |
Read GO annotations from a file in the GeneOntology format.
| [in] | fileName,the | name of the file containing the annotations. |
| [in] | symbolCol,an | optional additional column to consider for gene symbols. For example, sometimes the user is interested in all annotations of systematic gene id's. In the S. Cerevisiae Gene Ontology file, these are listed in Column 11. See the following link for details: http://www.geneontology.org/gene-associations/readme/sgd.README. Note that column 1 (not 0) indicates the first column. |
The method reads a file containing GO annotations in the standard 13-column GO format.
| void MyAnnotations::storeGODAG | ( | GeneOntology & | go | ) | [inline] |
Store a pointer to an instance of GeneOntology.
| void MyAnnotations::subtract | ( | MyAnnotations & | other, |
| MyAnnotations & | difference | ||
| ) |
Find the gene-function annotation pairs in the invocant that are not in other and store these pairs in difference.
| [in] | other,an | instance of MyAnnotations. |
| [out] | difference,an | instance of MyAnnotations that will contain the difference. |
 1.7.6.1
1.7.6.1

